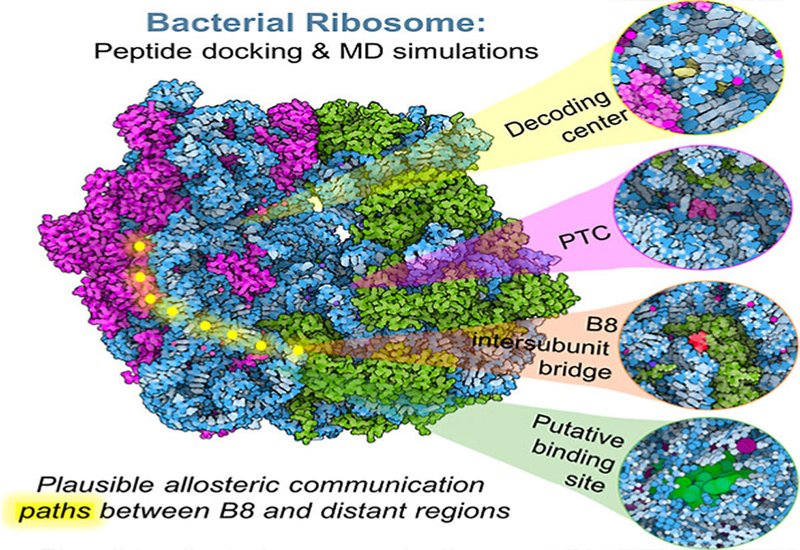
Antibiotic resistance has renewed urgency around the bacterial ribosome as a drug target, yet most discovery efforts focus on the two canonical orthosteric sites: the decoding center, DC, of the 30S small subunit and the peptidyl transferase center, PTC, of the 50S large subunit. Allosteric sites, particularly the intersubunit bridges that orchestrate subunit rotation and translational fidelity, remain underexplored, partly because the 2.5 MDa ribosomal complex resists conventional small-molecule virtual screening workflows. Peptide ligands offer a complementary strategy, given their capacity to engage large, charged RNA surfaces, but high-throughput docking against RNA targets is complicated by intrinsic RNA flexibility and high electronegativity. A computational framework capable of handling both orthosteric and allosteric pockets while ranking peptide candidates by mechanistic plausibility is therefore needed.
Researchers in the Sungur and Kurkcuoglu Groups at Istanbul Technical University, published in Biochemistry, developed an end-to-end pipeline that combines SiteMap-guided pocket detection, consensus docking with Glide and rDock, all-atom truncated molecular dynamics, MD, simulations, and coarse-grained MD, CGMD, simulations to screen roughly 23,000 conformers drawn from three peptide libraries: DBAASP v3, CyclicPepedia, and CycPeptMPDB. Four sites on the E. coli ribosome served as targets: the DC and PTC as orthosteric references, a putative allosteric pocket on the 30S subunit predicted by Gaussian Network Model analysis, and the B8 intersubunit bridge. Consensus scoring filtered candidates whose z-scores fell below −1.65 in both Glide and rDock, and top peptides advanced to three independent 100 ns all-atom MD simulations per site. A first application of the Desmond Viparr module to truncated ribosome-peptide complexes enabled accurate RNA force field parameterization across all four systems.
Docking and MD simulations identified peptides that recapitulated the hallmark contacts of the co-crystallized inhibitors viomycin at the DC and dalfopristin at the PTC. At the DC, candidates bearing the "DSF" sequence reproduced π−π stacking with G1491 and basic-group contacts with G1405 that mirror viomycin's crystallographic fingerprint. At the PTC, macrocyclic candidates presented aromatic surfaces favoring edge-face interactions with A2062 and A2451 while maintaining hydrogen bonds with G2505, reproducing the electrostatic-aromatic dual anchoring characteristic of dalfopristin. Interaction fingerprints were derived for each candidate by mapping contact types to the physicochemical class of the contacting peptide residue, providing site-specific scaffolding rules that rationalize binding affinity. CycPeptMPDB_2508, a cyclic peptide comprising Pro-Tyr-Mono83, achieved favorable docking scores and stable MD trajectories across all four sites, nominating it as a versatile lead core for further optimization.
Four independent 750 ns CGMD simulations of the apo 70S complex captured microsecond-scale equilibrium fluctuations and yielded a dynamic cross-correlation matrix with Pearson correlation coefficients of 0.74 for 16S rRNA and 0.75 for 23S rRNA when compared with experimental B-factors. Principal component analysis revealed that the first principal component captures the ratchet-like intersubunit rotation central to tRNA translocation. Residue interaction network analysis identified hub nucleotides with high betweenness centrality concentrated near the putative binding pocket and the B8 bridge, including G351, A356, and U367 on helices h14 and h15. A shortest-path analysis traced an allosteric communication route from the B8 bridge through the putative pocket, traversing h14, h15, ribosomal protein s4, and helix h16 to the 30S shoulder. Perturbation of U367 produced the largest reduction in allosteric coupling efficiency, identifying it as a functional bottleneck along that pathway. Hub nucleotides along this route showed greater than 98% sequence conservation, consistent with a functionally constrained signaling conduit.
The workflow offers three portable design principles for ribosome-targeted peptides. First, electrostatic-aromatic anchoring, stacking against purines combined with basic handles and directed hydrogen bonds, transfers across distinct ribosomal pockets and may guide scaffold optimization. Second, the shared contact chemistry across the DC, PTC, putative pocket, and B8 bridge supports a modular view of peptide-ribosome recognition in which consensus scaffolds could engage multiple pockets; CycPeptMPDB_2508 provides an initial structural proof of this concept. Third, the allosteric pathway map linking B8 to the 30S shoulder identifies leverage points for tuning decoding fidelity without occupying the primary antibiotic sites. The authors propose in vitro validation of prioritized peptides, targeted mutagenesis at network hubs, and iterative peptide redesign guided by the interaction fingerprints as immediate next steps toward experimental tractability.